Instruments Used



Overview
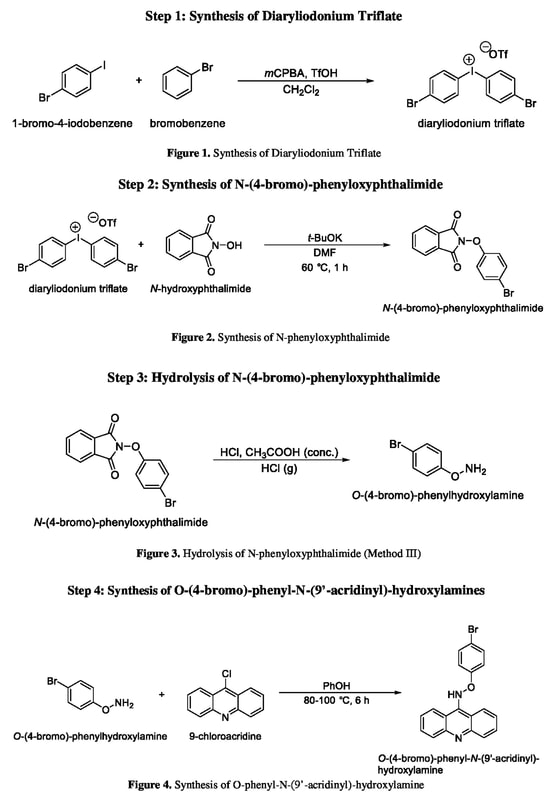
Synthesis of Diaryliodonium Triflate Salt
By using the procedures described by Bielawski, the triflate salt is able to be synthesized. A clean round bottom flask is stoppered and kept under inert atmosphere conditions by introducing argon gas. At this point 2mL of dichloromethane (DCM) were added to the flask followed by 116 mg (0.52 mmol, 1.1 equivalents) of mCPBA. After adding these, 54.43μL (0.52 mmol) of bromobenzene is added preceding 120μL (3.0 equivalency) of trifluoromethanesulfonic acid. Finally 130.13mg of 1-bromo-(4-iodo)-benzene is added to the mixture and it is stirred for 1 hour at room temperature. After 1 hour the solution is put in a rotary evaporator at 25 degrees Celsius to remove the solvent. To the mixture is then added 2mL of dietholether (Et2O) which is then stirred for 10 minutes. This process will force the impurities in the solution into the dietholether and the product to crystalize. The flask is left in the fridge for at atleast 30 minutes before removal. The solid was then washed with Et2O and rotary evaporated again to dry the diaryliodonium salt crystals before proceeding with the next step.
Synthesis of N-phenyloxyphthalimide
The methods to synthesize the N-phenyloxyphthalimide are outlined by Ghosh and are used in these procedures. In a new 25mL round bottom flask, 61.7mg of Potassium t-butoxide (t-BuOK) was dissolved in 2mL of anhydrous dimethyl formamide (DMF). Then 61.7mg of N-hydroxyphthalimide was added to the flask and the mixture was stirred for 10 minutes. If the compounds do not dissolve 2 more mL of DMF are added to the mixture before the triflate salts from the previous step are added. After adding the triflate salts, the flask is brought to 60 degrees Celsius using a variac and mantle and the mixture is stirred for 1 hour. After stirring was completed, the compound and solvent along with 20mL of deionized water and 20mL of ethelacetate are added to a seperatory funnel. The solution is shaken vigorously in order to mix the solution thoroughly and then the aqueous layer is drained into a waste beaker. 20 more mL of deionized water area added and the process is repeated three times. On the fourth time, rather than adding DI water, 20mL of brine was added, turning the solution a milky white color, to ensure the best separation of the organic and aqueous layer. The organic layer was then transferred to a 50mL Erlenmeyer flask which was stoppered. At this point added is anhydrous sodium sulfate in small increments. After each increment the flask is shaken vigorously to help absorb any remaining water in the solution. The increments were stopped when the anhydrous sodium sulfate stopped clumping together and started to look like a snow globe in the Erlenmeyer flask. Using gravity filtration, the solid was removed from the mixture and the remaining compounds were drained into a new 25mL round bottom flask. The flask was left under a fume hood allowing the ethelacetate to evaporate, leaving behind N-phenyloxyphthalimide.
Hydrolysis of N-phenyloxyphthalimide
Although there are many ways to send the N-phenyloxyphthalimide through hydrolysis, in this research the method outlined by Carlson where the compound was under acidic conditions was used. The compound in a 25mL round bottom from the previous step will be transferred to a 100mL round bottom flask to begin. The flask will be heated to 50 degrees Celsius using a variac and a heating mantle following which 15 mL of glacial acetic acid and 5 mL 37% hydrochloric acid (HCl) will be added. The solution will be heated to reflux, where the vapors turn to stock. At this point 1 mL of 37% HCl will be added to the round bottom flask periodically for 2 hours as it refluxes. After two hours the round bottom will be set out to cool to room temp. At this point the flask will be rotovapped to remove all acid and the remaining solid was scraped and transferred to a 50mL Erlenmeyer flask. To ensure the highest yields, the 100mL round bottom will be rinsed with DI water and added to the Erlenmeyer flask; the resulting solution should be no greater than 20mL. At this point in the hydrolysis the solution will be made alkaline by through the dropwise addition of 10% sodium hydroxide (NaOH). The new mixture will be transferred to a 125mL seperatory funnel along with 30mL of DCM and the aqueous layer was removed three times. After using the seperatory funnel, the compounds were dried by using anhydrous sodium sulfate after which gravity filtration was used to remove the solid. HCl was bubbled through the solution to ensure that no more precipitate is formed, and then let to stir for 30 minutes After being filtered using suction filtration, the solid is let to dry overnight.
Synthesis of O-(4-bromo)-phenyl-N-(9’-acridinyl)-hydroxylamines.
The final step in the synthesis of O-(4-bromo)-phenyl-N-(9’-acridinyl)-hydroxylamine will be based on the procedure outlined by Carlson. In a 100mL round bottom flask 3.29g of phenol will be heated to 88 degrees Celsius. At this point the O-phenylhydroxylamine synthesized in step 3 will then be added in a ratio of 1.5 mol O-phenylhydroxylamine: 1 mol 9-chloroacridine. This mixture will then be stirred at 80-100 °C for a period of 6 hours. After the mixture has been stirred it will be allowed to cool to room temperature. At this point 50mL of HCl is added. Next, the reaction mixture and a solution of 0.1 M sodium hydroxide (100 mL NaOH per 1 mol PhOH) will be added to a seperatory funnel to remove any excess phenol from the product. Once again the compound will be dried using anhydrous sodium sulfate after which the solid is removed via gravity filtration. The final product will be concentrated using the rotary evaporator. When the product has been fully concentrated and purified, it will be analyzed through the use of H-NMR and infrared (IR) spectroscopy.
By using the procedures described by Bielawski, the triflate salt is able to be synthesized. A clean round bottom flask is stoppered and kept under inert atmosphere conditions by introducing argon gas. At this point 2mL of dichloromethane (DCM) were added to the flask followed by 116 mg (0.52 mmol, 1.1 equivalents) of mCPBA. After adding these, 54.43μL (0.52 mmol) of bromobenzene is added preceding 120μL (3.0 equivalency) of trifluoromethanesulfonic acid. Finally 130.13mg of 1-bromo-(4-iodo)-benzene is added to the mixture and it is stirred for 1 hour at room temperature. After 1 hour the solution is put in a rotary evaporator at 25 degrees Celsius to remove the solvent. To the mixture is then added 2mL of dietholether (Et2O) which is then stirred for 10 minutes. This process will force the impurities in the solution into the dietholether and the product to crystalize. The flask is left in the fridge for at atleast 30 minutes before removal. The solid was then washed with Et2O and rotary evaporated again to dry the diaryliodonium salt crystals before proceeding with the next step.
Synthesis of N-phenyloxyphthalimide
The methods to synthesize the N-phenyloxyphthalimide are outlined by Ghosh and are used in these procedures. In a new 25mL round bottom flask, 61.7mg of Potassium t-butoxide (t-BuOK) was dissolved in 2mL of anhydrous dimethyl formamide (DMF). Then 61.7mg of N-hydroxyphthalimide was added to the flask and the mixture was stirred for 10 minutes. If the compounds do not dissolve 2 more mL of DMF are added to the mixture before the triflate salts from the previous step are added. After adding the triflate salts, the flask is brought to 60 degrees Celsius using a variac and mantle and the mixture is stirred for 1 hour. After stirring was completed, the compound and solvent along with 20mL of deionized water and 20mL of ethelacetate are added to a seperatory funnel. The solution is shaken vigorously in order to mix the solution thoroughly and then the aqueous layer is drained into a waste beaker. 20 more mL of deionized water area added and the process is repeated three times. On the fourth time, rather than adding DI water, 20mL of brine was added, turning the solution a milky white color, to ensure the best separation of the organic and aqueous layer. The organic layer was then transferred to a 50mL Erlenmeyer flask which was stoppered. At this point added is anhydrous sodium sulfate in small increments. After each increment the flask is shaken vigorously to help absorb any remaining water in the solution. The increments were stopped when the anhydrous sodium sulfate stopped clumping together and started to look like a snow globe in the Erlenmeyer flask. Using gravity filtration, the solid was removed from the mixture and the remaining compounds were drained into a new 25mL round bottom flask. The flask was left under a fume hood allowing the ethelacetate to evaporate, leaving behind N-phenyloxyphthalimide.
Hydrolysis of N-phenyloxyphthalimide
Although there are many ways to send the N-phenyloxyphthalimide through hydrolysis, in this research the method outlined by Carlson where the compound was under acidic conditions was used. The compound in a 25mL round bottom from the previous step will be transferred to a 100mL round bottom flask to begin. The flask will be heated to 50 degrees Celsius using a variac and a heating mantle following which 15 mL of glacial acetic acid and 5 mL 37% hydrochloric acid (HCl) will be added. The solution will be heated to reflux, where the vapors turn to stock. At this point 1 mL of 37% HCl will be added to the round bottom flask periodically for 2 hours as it refluxes. After two hours the round bottom will be set out to cool to room temp. At this point the flask will be rotovapped to remove all acid and the remaining solid was scraped and transferred to a 50mL Erlenmeyer flask. To ensure the highest yields, the 100mL round bottom will be rinsed with DI water and added to the Erlenmeyer flask; the resulting solution should be no greater than 20mL. At this point in the hydrolysis the solution will be made alkaline by through the dropwise addition of 10% sodium hydroxide (NaOH). The new mixture will be transferred to a 125mL seperatory funnel along with 30mL of DCM and the aqueous layer was removed three times. After using the seperatory funnel, the compounds were dried by using anhydrous sodium sulfate after which gravity filtration was used to remove the solid. HCl was bubbled through the solution to ensure that no more precipitate is formed, and then let to stir for 30 minutes After being filtered using suction filtration, the solid is let to dry overnight.
Synthesis of O-(4-bromo)-phenyl-N-(9’-acridinyl)-hydroxylamines.
The final step in the synthesis of O-(4-bromo)-phenyl-N-(9’-acridinyl)-hydroxylamine will be based on the procedure outlined by Carlson. In a 100mL round bottom flask 3.29g of phenol will be heated to 88 degrees Celsius. At this point the O-phenylhydroxylamine synthesized in step 3 will then be added in a ratio of 1.5 mol O-phenylhydroxylamine: 1 mol 9-chloroacridine. This mixture will then be stirred at 80-100 °C for a period of 6 hours. After the mixture has been stirred it will be allowed to cool to room temperature. At this point 50mL of HCl is added. Next, the reaction mixture and a solution of 0.1 M sodium hydroxide (100 mL NaOH per 1 mol PhOH) will be added to a seperatory funnel to remove any excess phenol from the product. Once again the compound will be dried using anhydrous sodium sulfate after which the solid is removed via gravity filtration. The final product will be concentrated using the rotary evaporator. When the product has been fully concentrated and purified, it will be analyzed through the use of H-NMR and infrared (IR) spectroscopy.

